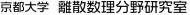
化学グラフの推定問題に対するアルゴリズムの研究
新規化合物の設計は薬学,医学,農学などにおいて重要であり,ポストゲノム時代における生体生命情報学の主要目標の一つです. このプロジェクトでは,新規化学構造を計算機により導き出す計算手法について, 理論基盤構築および実用的アルゴリズムの両面から研究します.
新規化合物の設計は薬学,医学,農学などにおいて重要であり,ポストゲノム時代における生体生命情報学の主要目標の一つです. そのために様々な情報解析手法が研究されてきましたが,その一つに化学構造とその活性の関係を解析・推定する「構造活性相関」があります. この構造活性相関について,近年,様々な機械学習が応用されるようになってきました. 特に,サポートベクターマシンを用いる方法では,下の図1に示すように,通常,対象となるデータを,有限次元のユークリッド空間,もしくは,無限次元のヒルベルト空間の点(特徴ベクトルとよ ばれる)に対応付ける写像を定義し,写像された空間における超平面を用いることにより,分類や予測を行います. 活性予測のためには,化学構造を特徴ベクトルに変換する必要がありますが,通常,化学構造はグラフ構造として入力され,そのグラフが特徴ベクトルに変換されます.
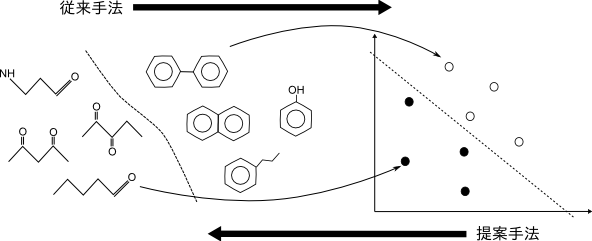
本研究は,従来手法の逆を行うことにより新規化学構造を計算機により導き出す計算手法について研究します. 具体的には,「特徴ベクトルから,もとの化学構造を推定する」ことにより新規な化学構造を導き出す方法について,理論基盤構築および実用的アルゴリズムの両面から研究します.この方法が開発できると様々な応用の可能性があります. 例えば,化合物Aと化合物Bの中間の性質を持つ化合物Cを設計したいとしましょう(下図参照). この場合,Aの特徴ベクトルとBの特徴ベクトルの中点を計算し,それを逆写像することにより中間の性質を持つと期待される化合物Cの構造を得ることができます. 本研究のアイデアや成果を数年で薬剤設計に応用することは難しいですが,十年,もしくは,二十年先の薬剤設計に役立つ可能性はあると考えています.
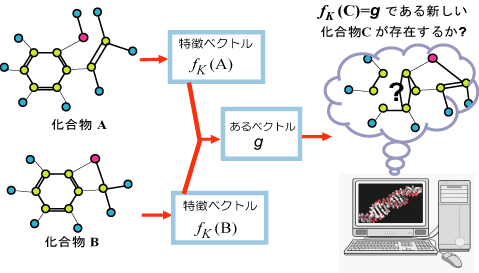
また,特徴ベクトルからの推定以外に,NMRや質量分析スペクトルからの構造推定に本研究の成果を活かすこともできると考えています. このような分析装置は与えられた試料の化学組成,一部の細かな構造(フラグメント)に関する情報を提供しますが,試料の化学構造が未知の場合,これらの情報に適合する化学構造を推定する問題を解くことが必要になります.
具体的には以下を本研究の目標とします.
- グラフの逆写像問題の計算論的側面について現在の研究をより深化させ,どのような種類のグラフ,どのような種類の特徴ベクトルであれば,厳密解,もしくは,近似解が理論的に効率よく(多項式時間で)計算や数え上げができるのかを明らかにする.
- 特徴ベクトルから化学構造に対応するグラフ構造を実用的な時間で計算するためのアルゴリズムを開発する. この場合,特徴ベクトル以外にも,化合物が備えるべき制約条件を入力できるようにする.なお,既に一般的な場合における計算困難性(NP困難性)が示されているため,できるだけ大きなサイズの化合物に対して実用的な時間で厳密解もしくは(精度保証のある)近似解を計算できるようにする.具体的には(水素原子を除いて)50〜100原子程度の化合物まで対応することを目標とする.
- 開発したアルゴリズムを実際に利用可能とする WEB サーバーを作成・公開し,商業目的以外にはフリーで利用できるようにする.
本研究で開発する手法は,様々な制約下で可能な構造を数え上げることもできるようにする予定ですので,それらをツールとして活用することにより,思いもしなかった化学構造が提示され,化学者の研究に役立つ可能性があります. 例えば,フラーレン(C60)の構造はいくつかの制約から最終的には化学者が自分の頭で導いたものですが,そのような発見を計算機により行えるようになる可能性があります.
現在,特徴ベクトルの与え方にもよるが,木形状であれば25原子程度(水素原子を除く)の化合物の連結構造をすべて列挙することができています[1].提案法はアルカン(CnH2n+2)の列挙に特化した場合でも従来法に比べ高速で,n=31のすべてのアルカンを列挙できました.このアルゴリズムはどなたでも利用頂けるように阿久津研究室との協力のもとウエブサーバーとして以下のサイト公開しております.
http://sunflower.kuicr.kyoto-u.ac.jp/tools/enumol2/
[1] H. Fujiwara, W. Jiexun, L. Zhao, H. Nagamochi, T. Akutsu, Enumerating tree-like chemical graphs with given path frequency, Journal of Chemical Information and Modeling, 48 1345-1357, 2008.